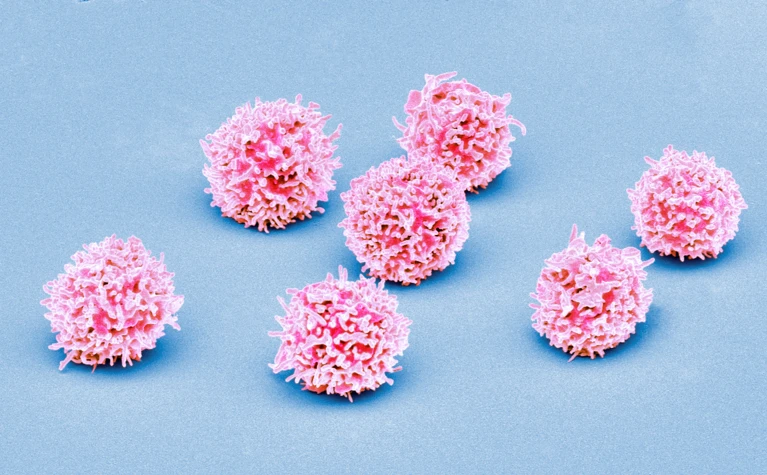
Normal T cells (artificially coloured) can be crowded out by the abnormal immune cells characteristic of T-cell acute lymphoblastic leukaemia.Credit: Steve Gschmeissner/Science Photo Library
An aggressive blood cancer that mainly affects children has 15 distinct subtypes, each linked to a particular outcome and responsiveness to drugs, according to a genomic analysis1. The work holds promise for improving treatment — for example, sparing some children the harshest chemotherapy regimens and giving others the latest immune therapies.
This detailed classification paves the way for targeted therapies, researchers say, offering hope for people with T-cell acute lymphoblastic leukaemia (T-ALL), which comprises roughly 5% of all paediatric cancers. The work, published today in Nature, might help to predict who is less likely to respond to treatment, and it could guide doctors in selecting more-effective therapies from the start.
“It’s a wonderful study that will be a very rich resource for everybody … treating T-ALL patients,” says Jan Cools, a researcher in leukaemia genetics at the Flemish Institute for Biotechnology in Ghent, Belgium, who was not involved in the research.
A stem cell gone bad
T-ALL occurs when a mutant stem cell in the bone marrow churns out large numbers of abnormal T-cells, a type of immune cell. Although survival rates for T-ALL have improved with advances in chemotherapy, 15–20% of children and teenagers experience relapses or have forms of the disease that don’t respond to standard treatment, says study co-author David Teachey, a paediatric oncologist and researcher at the Children’s Hospital of Philadelphia in Pennsylvania. That means it’s important to find better biological markers that can predict which people with T-ALL will need targeted therapies or new treatment approaches, he says.
Previous research had identified various subtypes of T-ALL, but no study had been large enough to reliably predict a person’s disease course on the basis of genetic changes alone. So, Teachey and his colleagues analysed the entire DNA sequence of both tumour cells and healthy cells from more than 1,300 people with T-ALL who received the same treatment. The researchers also examined cellular RNA to understand how gene activity was altered in cancer samples.
Linking genome and outcome
The analysis uncovered 15 distinct T-ALL subtypes, some previously uncharacterized. Each subtype showed unique genetic alterations and gene-expression patterns. People with certain subtypes were more likely to have cancer cells remaining in their body after treatment, which can lead to a relapse of the disease. People who had other subtypes were more likely to survive cancer-free, and one subtype was more likely to lead to another type of cancer elsewhere in the body, the researchers found.
The analysis also revealed that nearly 60% of genetic changes associated with T-ALL occur in stretches of DNA that do not produce proteins but can influence gene activity. These alterations often caused inappropriate activation of genes, contributing to cancer development.
Could CAR-T-cell therapy offer hope to children with cancer?
Using genetic and clinical data, the researchers classified T-ALL according to risk level: very high, high, low and very low. This classification might help doctors to customize treatments by recommending stronger chemotherapy or new immune therapies for high-risk individuals and less aggressive treatments for those at lower risk, Cools says.
The study drew participants from the United States, Canada, Australia, Switzerland and New Zealand. Since a person’s genetic background can influence their response to treatment, the findings need to be validated across diverse populations, says study co-author Charles Mullighan, a haematologist at St. Jude Children’s Research Hospital in Memphis, Tennessee.
The research also emphasizes the need to analyse the entire DNA sequence of tumour cells in people with T-ALL. Although this ‘whole-genome sequencing’ is not yet widespread owing to its cost, Mullighan expects it to become more common in the future. “It’s studies like this that make a solid case that we should be increasingly performing whole-genome sequencing for these types of leukaemia.”